Protein Crystallization & Protein Crystallography Services
A Brief History of Protein Crystallography
Protein crystallization and protein crystallography is the oldest method for structure determination at atomic resolution. It won't be an exaggeration to state that this method created the foundation of structural biology and provided valuable and deep insights into the mechanisms of function of organisms at the molecular level. Various areas of biological sciences benefited from structural insights, and among these is, of course, drug discovery.
The history of X-ray crystallography started with the discovery of X-rays by Conrad Röntgen (Nobel Prize in Physics, 1901). The electromagnetic nature of X-rays and the physical principles of X-ray diffraction were discovered later by Max von Laue (Nobel Prize in Physics, 1914), while the foundation for X-ray crystallography was laid by William Henry Bragg and William Lawrence Bragg (father and son, Nobel Prize in Physics, 1915). Later, starting from determining the first protein crystal structure (Nobel Prize to Max Perutz & John Kendrew, 1962), many Nobel Prizes were awarded to discoveries based on protein crystallography.
CROs providing protein crystallization and protein crystallography services are a relatively new phenomenon. This became possible after the impressive developments in the 1990-ties, which made protein structure determination much more efficient and affordable. SARomics Biostructures, since its foundation in 2006, has served the community and accumulated broad experience in handling different structural biology projects.
Here we give an overview of the typical workflow of our protein crystallization and protein crystallography services. The experiments include but are not limited to gene-to-structure X-ray crystallography, antibody and antibody-antigen crystallization and structure analysis, protein-ligand crystallization and structure determination. You may contact us through the contact form to inquire about our services.
The history of X-ray crystallography started with the discovery of X-rays by Conrad Röntgen (Nobel Prize in Physics, 1901). The electromagnetic nature of X-rays and the physical principles of X-ray diffraction were discovered later by Max von Laue (Nobel Prize in Physics, 1914), while the foundation for X-ray crystallography was laid by William Henry Bragg and William Lawrence Bragg (father and son, Nobel Prize in Physics, 1915). Later, starting from determining the first protein crystal structure (Nobel Prize to Max Perutz & John Kendrew, 1962), many Nobel Prizes were awarded to discoveries based on protein crystallography.
CROs providing protein crystallization and protein crystallography services are a relatively new phenomenon. This became possible after the impressive developments in the 1990-ties, which made protein structure determination much more efficient and affordable. SARomics Biostructures, since its foundation in 2006, has served the community and accumulated broad experience in handling different structural biology projects.
Here we give an overview of the typical workflow of our protein crystallization and protein crystallography services. The experiments include but are not limited to gene-to-structure X-ray crystallography, antibody and antibody-antigen crystallization and structure analysis, protein-ligand crystallization and structure determination. You may contact us through the contact form to inquire about our services.
Preparing a protein for crystallization
For crystallization, it is essential to get a sufficient amount of high-purity protein, also called crystallization-grade protein. Our services include recombinant protein expression and purification (see also our catalog of high purity crystallization grade proteins) adapted for obtaining high-purity and stable crystallization-grade proteins.
After cloning, expression, and purification, and before crystallization, an accurate biophysical characterization of the state of the recombinant protein in solution is performed. The purified protein in the solution must be well-folded, stable, and monodisperse at a given concentration and pH range. This means that the solution should not contain any denatured and aggregated material. This may be assessed, for example, by dynamic light scattering (DLS), which will quickly reveal the presence of any aggregated material.
Our services also include the following methods for the biophysical characterization of the protein:
We use protein NMR spectroscopy to know whether the construct results in a folded, well-behaved protein. The HSQC fingerprint spectrum of the protein readily shows whether the protein is well-folded, unfolded, or in a ‘molten-globule’ state and if some parts of the protein are flexible.
The service protocols for characterization and crystallization have been developed to accelerate the project depending on the type of protein.
After cloning, expression, and purification, and before crystallization, an accurate biophysical characterization of the state of the recombinant protein in solution is performed. The purified protein in the solution must be well-folded, stable, and monodisperse at a given concentration and pH range. This means that the solution should not contain any denatured and aggregated material. This may be assessed, for example, by dynamic light scattering (DLS), which will quickly reveal the presence of any aggregated material.
Our services also include the following methods for the biophysical characterization of the protein:
- DLS (Dynamic Light Scattering)
- CD spectroscopy
- DSF, Differential Scanning Fluorimetry (also called thermal shift assay)
- Protein NMR spectroscopy
We use protein NMR spectroscopy to know whether the construct results in a folded, well-behaved protein. The HSQC fingerprint spectrum of the protein readily shows whether the protein is well-folded, unfolded, or in a ‘molten-globule’ state and if some parts of the protein are flexible.
The service protocols for characterization and crystallization have been developed to accelerate the project depending on the type of protein.


The plate hotel on the image is part of our technology platform and is used for storing and monitoring crystallization plates, which are kept at a constant temperature.
The second image shows a 96-well crystallization plate from Hampton Research
The second image shows a 96-well crystallization plate from Hampton Research
Factors Affecting Protein Crystallization
Together with protein characterization, our services include a develop a crystallization protocol for obtaining well-diffracting single crystals for protein crystallographic structure determination. The number of experimental parameters affecting protein crystallization can be huge. The most common protocols rely on the variation of the following parameters:
* Type of buffer and its pH
* Ionic strength
* The presence of various salts in the solution
* The presence of ligands (co-factors, substrate analogs, inhibitors)
* The type of precipitant used (polyethylene glycol (PEG) and ammonium sulfate are the most common)
Many conditions must be screened before a successful crystallization protocol can be formed. Hundreds and often thousands of conditions are tested until "good" crystallization conditions are identified. Commercial screens, like those from Hampton Research or Molecular Dimensions, are initially used.
The most common method for water-soluble protein crystallization is sitting or hanging drops. High throughput and high precision liquid handling and imaging robotics are also required for the best efficiency of the crystallization efforts. For example, 96 conditions can be screened using robotics with as little as 15 microL of protein sample. Our services platform has liquid-handling robots to design screens for optimizing crystallization conditions identified in the initial trials. This helps in saving both time and precious material. The crystallization plates are stored in plate hotels at a constant temperature to avoid temperature fluctuations in laboratory conditions.
Our guide for shipping samples may be helpful for information on handling and sending samples. Of course, you may inquire directly about the details of our services using the contact form.
* Type of buffer and its pH
* Ionic strength
* The presence of various salts in the solution
* The presence of ligands (co-factors, substrate analogs, inhibitors)
* The type of precipitant used (polyethylene glycol (PEG) and ammonium sulfate are the most common)
Many conditions must be screened before a successful crystallization protocol can be formed. Hundreds and often thousands of conditions are tested until "good" crystallization conditions are identified. Commercial screens, like those from Hampton Research or Molecular Dimensions, are initially used.
The most common method for water-soluble protein crystallization is sitting or hanging drops. High throughput and high precision liquid handling and imaging robotics are also required for the best efficiency of the crystallization efforts. For example, 96 conditions can be screened using robotics with as little as 15 microL of protein sample. Our services platform has liquid-handling robots to design screens for optimizing crystallization conditions identified in the initial trials. This helps in saving both time and precious material. The crystallization plates are stored in plate hotels at a constant temperature to avoid temperature fluctuations in laboratory conditions.
Our guide for shipping samples may be helpful for information on handling and sending samples. Of course, you may inquire directly about the details of our services using the contact form.


The first image shows drops with protein crystals, while the second shows a good-quality protein crystal diffraction. Protein crystallography uses the intensities of the spots measured from the diffraction images to calculate the electron density map into which the protein structure model is built.
Bringing the Crystal to the X-ray Beam
Protein crystallography requires good-quality crystals. When such crystals are obtained, it is time for the first test in an X-ray beam. Then, if X-ray diffraction from the crystals is of good quality, data are collected. For our services, X-ray data collection is run at the MAX IV synchrotron radiation facility a few kilometers from our Lund labs. Alternatively, we send the crystals to other European synchrotrons.
For protein crystallographic structure determination, we need phase information. When an experimental structure of the same protein is already available, the molecular replacement method is applied to obtain phase information and subsequent calculation of an electron density map into which the protein model will be built. Sometimes is also possible to use models generated by the AlphaFold project for an initial phasing. Molecular replacement will substantially accelerate the protein crystallography project. When no reliable model is available, various experimental methods may be used for obtaining phase information. Further refinement of the model will ensure that the fine details of the structure (accurate positions of protein and ligand atoms) are well resolved in the electron density map. Our services platform is well equipped for running these processes time-efficiently.
Our educational site provides additional practical information on protein crystallography, including crystallization and structure determination.
For protein crystallographic structure determination, we need phase information. When an experimental structure of the same protein is already available, the molecular replacement method is applied to obtain phase information and subsequent calculation of an electron density map into which the protein model will be built. Sometimes is also possible to use models generated by the AlphaFold project for an initial phasing. Molecular replacement will substantially accelerate the protein crystallography project. When no reliable model is available, various experimental methods may be used for obtaining phase information. Further refinement of the model will ensure that the fine details of the structure (accurate positions of protein and ligand atoms) are well resolved in the electron density map. Our services platform is well equipped for running these processes time-efficiently.
Our educational site provides additional practical information on protein crystallography, including crystallization and structure determination.
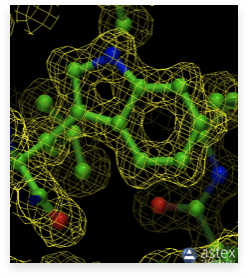
An essential requirement of protein crystallography is a well-resolved (good resolution) electron density map. An example of such a map showing the side chain of tryptophan built into its density is shown in the image.