
Fragment Library Screening & Lead Discovery Services
Hit identification, hit to lead & lead optimization services
SARomics Biostructures' fragment screening, structure-based lead discovery, and lead optimization services offer access to our proprietary weak-affinity chromatography (WAC™) screening technology. Our services include:
Our hit-to-lead & lead optimization services are guided by protein X-ray crystallographic analysis of the target structure in complex with the compounds of interest.
WAC™ is a robust and cost-efficient screening method. It can be combined with biophysical screening techniques for hit identification and verification, such as thermal-shift assay (DSF), isothermal titration calorimetry (ITC), and protein NMR spectroscopy. SARomics Biostructures and our partner Red Glead Discovery jointly own the commercial rights of the WAC™ screening technology.
Different drug discovery strategies can be applied depending on the availability of structural information. This is briefly discussed in our recent blog post and with more details on our technology pages. You may also contact us to discuss the details of our services and your project.
For details of the services, please follow the links below.
- Fragment screening & hit identification using WAC™ screening technology (proprietary fragment library or custom compound screening library)
- Structure-based hit to lead and lead optimization services
- Computational Chemistry services: pharmacophore and shape-based virtual screening services, scaffold hopping, design of screening library, QSAR analysis, and compound optimization
- Biochemical and cell-based assay and in vitro ADME
Our hit-to-lead & lead optimization services are guided by protein X-ray crystallographic analysis of the target structure in complex with the compounds of interest.
WAC™ is a robust and cost-efficient screening method. It can be combined with biophysical screening techniques for hit identification and verification, such as thermal-shift assay (DSF), isothermal titration calorimetry (ITC), and protein NMR spectroscopy. SARomics Biostructures and our partner Red Glead Discovery jointly own the commercial rights of the WAC™ screening technology.
Different drug discovery strategies can be applied depending on the availability of structural information. This is briefly discussed in our recent blog post and with more details on our technology pages. You may also contact us to discuss the details of our services and your project.
For details of the services, please follow the links below.

At the center of our fragment screening & lead discovery services is the high throughput (2000-3000 cmpds/week) proprietary weak-affinity chromatography (WAC™) screening technology, which provides high-quality data and has been validated against NMR spectroscopy and X-ray crystallography.
Our fragment library contains a collection of 1300 low-MW (<220) MedChem-friendly compounds (also available as neat samples, DMSO and DMSO-d6 solutions), designed to be general-purpose (not target-directed) covering diverse chemical space. Custom compound libraries can also be run.

Computation chemistry is an integral part of structure-based lead discovery. Our team has gathered extensive experience in computational chemistry and may provide a wide range of services, including:
- Virtual screening services
- Pharmacophore and shape-based virtual screening
- Ligand-based drug design, etc.
WAC™ screening technology
WAC™ Facts
- Screening and binding assay in one experiment
- Measure fragment affinity to immobilized protein
- Built-in quality control (MS)
Key Advantages
- Robust and accurate (validated against NMR and X-ray)
- Quick set-up and workflow (3 weeks turnaround)
- High throughput (>5000 cmpds/week)
- Low material consumption (<5 mg protein)
- Find mM hits by screening fragments at low concentrations (1-5 μM)
- Hit rates from 1% to 20%, (avg 6%)
Applications
- Screen for novel chemical starting points
- Assess the druggability of new targets
- Find differentiated backups in mature projects
- Rescue mode for challenging targets (PPI etc.)
Basics of WAC™ screening technology
WAC™ screening technology is the primary compound screening method used within our lead discovery services program. It is based on covalent immobilization of the protein target on a standard high-performance liquid chromatography (HPLC) column. A solution of small molecular weight fragments or larger compounds is injected into the column. During elution, the fragments with a higher affinity for the protein will stay on the column longer than those with a low affinity. The fragments can be conveniently detected using mass- or UV spectrometry. This method is an efficient and lower-cost choice compared to other biophysical screening methods used for compound screening, e.g., X-ray crystallography, protein NMR spectroscopy, or isothermal titration calorimetry (ITC).
After the initial hit identification, protein NMR spectroscopy or X-ray crystallography can be used to gain additional insights into the details of the interactions of a compound with the protein. Ligand efficiency and ligand binding energy may also be estimated to guide the hit-to-lead and lead optimization processes.
The commercial rights of the WAC™ screening technology are jointly owned by SARomics Biostructures and our partner Red Glead Discovery.
After the initial hit identification, protein NMR spectroscopy or X-ray crystallography can be used to gain additional insights into the details of the interactions of a compound with the protein. Ligand efficiency and ligand binding energy may also be estimated to guide the hit-to-lead and lead optimization processes.
The commercial rights of the WAC™ screening technology are jointly owned by SARomics Biostructures and our partner Red Glead Discovery.
Proprietary fragment library
Our fragment library includes a collection of 1300 compounds (available as neat samples, DMSO, and DMSO-d6 solutions).
Designed to be general-purpose (not target-directed), covering diverse chemical space.
A focus on low-molecular-weight fragments (<220)
Solubility data (DMSO, water) are available for >80% of the fragments
Analytical data (LCMS, 1H-NMR) available for all fragments
Designed to be MedChem friendly (HBDs, HBAs and ring count, ClogP, functional groups, etc.)
More than 90% commercially available, facilitating rapid SAR-generation

On the image: Klara Jonasson, PhD,
Senior Research Scientist, Red Glead Discovery

Computational chemistry & virtual screening services
Computational chemistry methods can be efficiently used in lead discovery, and they are essential in virtual screening and structure-based lead design. The process provides a combination of pharmacophore and shape-based virtual screening of fragments or large compound libraries, ligand docking, scaffold hopping, QSAR analysis, and ligand optimization, allowing substantial acceleration of the lead discovery process. Our virtual library used for in silico compound screening contains millions of purchasable molecules. It has already been pre-filtered to remove, e.g., reactive groups and compounds that are too lipophilic.
Our computational chemistry team collaborates closely with other teams to efficiently drive the project from hit identification, hit to lead, lead optimization, and candidate drug nomination. Our learning center discusses the details of various lead design strategies, which can be followed depending on the type of information at hand.
Our computational chemistry team collaborates closely with other teams to efficiently drive the project from hit identification, hit to lead, lead optimization, and candidate drug nomination. Our learning center discusses the details of various lead design strategies, which can be followed depending on the type of information at hand.
Our computational chemistry and virtual screening services include (but are not limited to) the following options:
- Pharmacophore and shape-based fragment screening
- Ligand-based drug design
- Scaffold hopping
- Design of screening library
- QSAR analysis and optimization

As part of our computational chemistry services, we also offer in silico protein optimization and structure-based design of biological drugs (biologics) and peptides using quantitative structure-activity relationships (QSAR) methodology as implemented in our proprietary ProPHECY™ software package.

On the image: Bo Svensson, PhD, Director, in silico discovery
Recent publications with a contribution by the SARomics team
Zetterberg FR, Diehl C, Håkansson M, Kahl-Knutson B, Leffler H, Nilsson UJ, Peterson K, Roper JA & Slack RJ (2023).
Discovery of Selective and Orally Available Galectin-1 Inhibitors
J. Med. Chem. https://doi.org/10.1021/acs.jmedchem.3c01787
PDB entries:
8OJP - Human galectin 1 in complex with inhibitor (on hold until publication)
A new series of orally available α-d-galactopyranosides with high affinity and specificity toward galectin-1 have been discovered. High affinity and specificity were achieved by changing six-membered aryl-triazolyl substituents in a series of recently published galectin-3-selective α-d-thiogalactosides (e.g., GB1107 Kd galectin-1/3 3.7/0.037 μM) for five-membered heterocycles such as thiazoles. One compound, GB1490 (Kd galectin-1/3 0.4/2.7 μM), was selected for further characterization toward a panel of galectins showing a selectivity of 6- to 320-fold dependent on galectin. The X-ray structure of GB1490 bound to galectin-1 reveals the compound bound in a single conformation in the carbohydrate binding site.
Discovery of Selective and Orally Available Galectin-1 Inhibitors
J. Med. Chem. https://doi.org/10.1021/acs.jmedchem.3c01787
PDB entries:
8OJP - Human galectin 1 in complex with inhibitor (on hold until publication)
A new series of orally available α-d-galactopyranosides with high affinity and specificity toward galectin-1 have been discovered. High affinity and specificity were achieved by changing six-membered aryl-triazolyl substituents in a series of recently published galectin-3-selective α-d-thiogalactosides (e.g., GB1107 Kd galectin-1/3 3.7/0.037 μM) for five-membered heterocycles such as thiazoles. One compound, GB1490 (Kd galectin-1/3 0.4/2.7 μM), was selected for further characterization toward a panel of galectins showing a selectivity of 6- to 320-fold dependent on galectin. The X-ray structure of GB1490 bound to galectin-1 reveals the compound bound in a single conformation in the carbohydrate binding site.
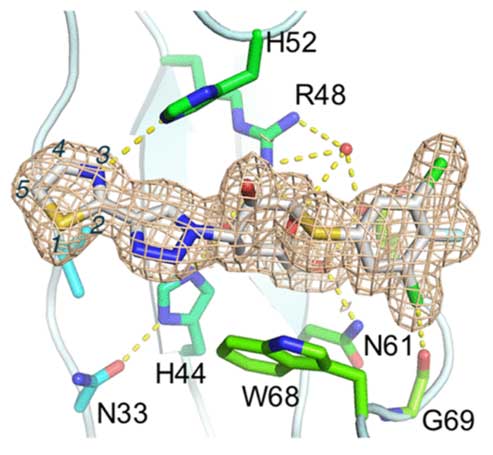
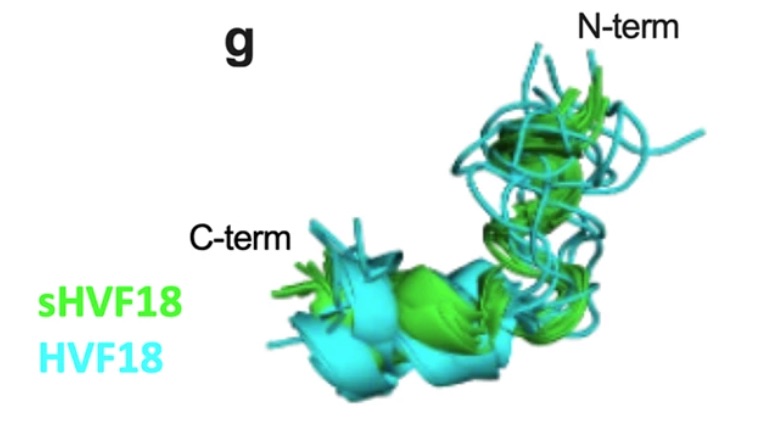
Petruk G, Puthia P, Samsudin F, Petrlova J, Olm F, Mittendorfer M, Hyllén S, Edström D, Strömdahl A-C, Diehl C, Ekström S, Walse B, Kjellström S, Bond PJ, Lindstedt S & Schmidtchen A (2023).
Targeting Toll-like receptor-driven systemic inflammation by engineering an innate structural fold into drugs.
Nat Commun, 14, 6097. https://doi.org/10.1038/s41467-023-41702-y
PDB entries:
8BWW - Targeting Toll-like receptor-driven systemic inflammation by engineering an innate structural fold into drugs
Thrombin-derived C-terminal peptides (TCPs) are endogenous anti-infective immunomodulators interfering with CD14-mediated TLR-dependent immune responses. Using a combination of structure- and in silico-based design, nuclear magnetic resonance (NMR) spectroscopy, biophysics, mass spectrometry, cellular, and in vivo studies, the paper presents the structure, CD14 interactions, protease stability, transcriptome profiling, and therapeutic efficacy of sHVF18.
Targeting Toll-like receptor-driven systemic inflammation by engineering an innate structural fold into drugs.
Nat Commun, 14, 6097. https://doi.org/10.1038/s41467-023-41702-y
PDB entries:
8BWW - Targeting Toll-like receptor-driven systemic inflammation by engineering an innate structural fold into drugs
Thrombin-derived C-terminal peptides (TCPs) are endogenous anti-infective immunomodulators interfering with CD14-mediated TLR-dependent immune responses. Using a combination of structure- and in silico-based design, nuclear magnetic resonance (NMR) spectroscopy, biophysics, mass spectrometry, cellular, and in vivo studies, the paper presents the structure, CD14 interactions, protease stability, transcriptome profiling, and therapeutic efficacy of sHVF18.
For related services please follow the respective link
Gene-to-structure services

Protein NMR spectroscopy

Antibody structure

Structure-based design of biological drugs