Biosimilars: Structure Comparability Analysis
In the second part of our article series on antibody therapeutics, we discuss biosimilars and their structure comparability analysis.
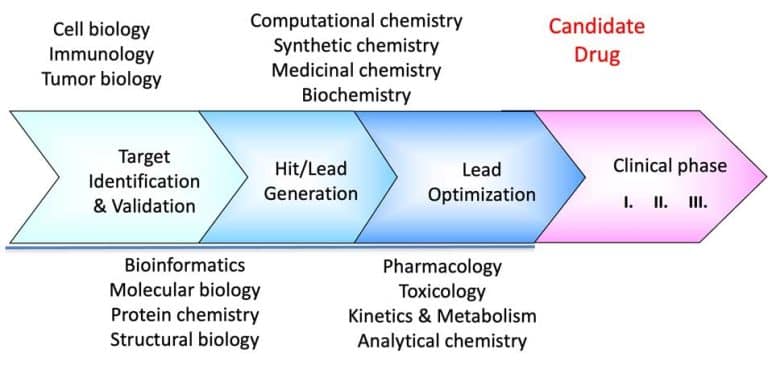
In the previous post, we discussed the general contribution of structural biology, particularly X-ray crystallography, to our knowledge in the field of monoclonal antibody engineering and the design of therapeutic antibodies. Here, we focus on biosimilar structure comparability analysis.
Biologics are generally large molecules, mainly proteins, produced by living cells. Their structures are considerably more complex than those of small-molecule drugs. As noted in our previous post, monoclonal antibodies constitute the largest group of biologics. Thus, in an analysis of the contribution of X-ray crystallography to drug discovery and design for 2010-2016, Westbrook & Burley (2019) identified 210 PDB entries describing FDA-approved drugs. Of those, 36 were biologics, with 24 being monoclonal antibodies. Currently, according to the Antibody Therapies Database, there are over 9400 monoclonal antibodies targeting over 2400 human disease conditions such as psoriasis, Crohn’s disease, ulcerative colitis, arthritis, kidney conditions, diabetes, and cancers (such as breast, lung, and colon).
The first monoclonal antibody therapeutic targeting CD3, a cell-surface protein complex with a critical role in T-cell biology, was approved in 1986 by the FDA. The antibody was developed by Stuart Schlossman and colleagues and later named muromonab-CD3. It entered the clinic as an immunosuppressive for the prevention of transplant rejection. Thirty-five years later, in 2021, the FDA approved the 100th therapeutic monoclonal antibody. In Europe, the first antibody therapy was approved in 1991 (withdrawn in 1993), and the second in 1995. Japan and China have been lagging after the USA and Europe but are rapidly catching up. The number of approved therapeutic monoclonal antibodies in different countries is also reflected in the number of companies involved in this business. As of 2022, of the 82 companies with at least one approved therapeutic antibody, 33 had drugs approved by the FDA, 15 by the European EMA, 17 by the NMPA (Chinese regulator), and seven by the Japanese regulator PDMA (see the global landscape of approved antibody therapies).
Biosimilars
Considering the time interval, we may expect several therapeutic antibodies already have or are coming out of patent in the next few years. And as with small-molecule drugs, pharmaceutical companies are racing to develop their follow-on variants, so-called biosimilars. Currently, 93 biosimilars have been approved by EMA for use in the EU, of which 38 are monoclonal antibodies. FDA has approved 41 biosimilars, of which 27 are monoclonal antibodies.
Unlike generic small-molecule drugs, which are synthesized in the laboratory and are exact copies of the reference drug, monoclonal antibodies are produced in mammalian cells due to the ability of these cells to perform post-translational modification. A drawback of expression in heterologous systems is unpredictable modifications like variable glycosylation, deamination of asparagine and glutamine, and oxidation of cysteine, methionine, or tryptophan, which result in heterogeneity in the biologic product. Unexpected modifications may affect the product’s chemical properties, such as surface charge, solubility, stability, and three-dimensional structure (higher order structure or HOS). These, in turn, may affect the efficacy and safety of the treatment. For this reason, biosimilars cannot be handled as generic small-molecule drugs (and they are not considered “generic drugs”). For approval by regulatory agencies, developers must thus present detailed comparative studies of the biosimilar, which has to be highly similar to the reference antibody, with no clinical differences in safety, quality, and efficacy, as stated by EMA. Among these parameters, biosimilar structure comparability analysis plays a central role.
The American FDA recommends using a stepwise process to prove biosimilarity “without the need to establish the safety and effectiveness of the proposed product independently.” The process is divided into 1) biosimilar structural analysis, 2) functional analysis, 3) animal data, and 4) clinical studies. First, extensive structural and functional characterization and comparison of the proposed and reference products should be performed. The guidelines stress, “The more comprehensive and robust the comparative structural and functional characterization — the more useful such characterization will be in determining what additional studies may be needed.” In addition, the FDA stresses that “appropriate analytical methodologies with adequate sensitivity and specificity for structural characterization of the proteins should be used.”
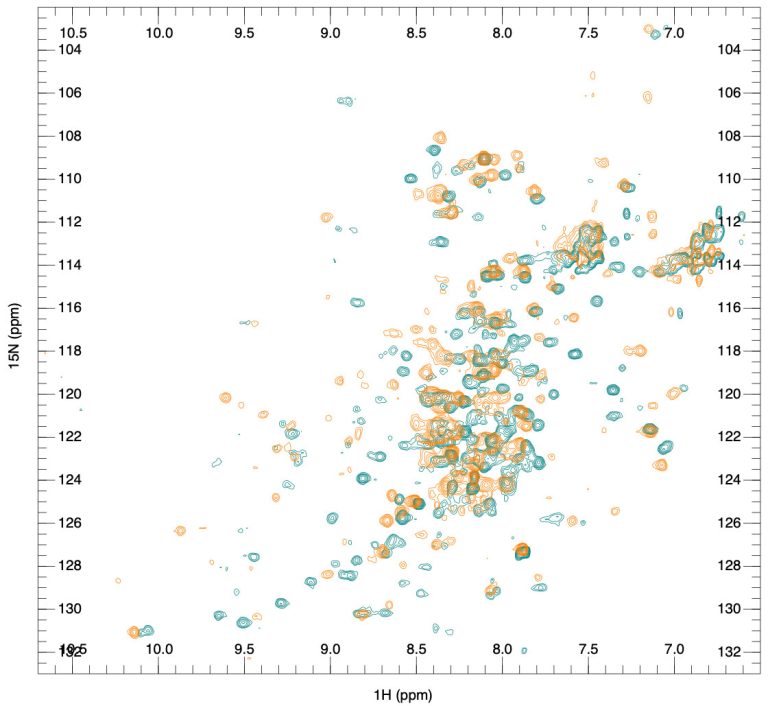
Biosimilar structure comparability analysis should describe the primary (amino acid sequence) and higher order structures (HOS), including secondary, tertiary, and quaternary structures. The EMA stresses the importance of comparability of the biosimilar’s and the originator antibody’s HOS. Undoubtedly, detailed structural characterization is highly relevant to the process. This is understandable, considering the importance of the molecule’s overall conformation, especially the conformation of the compatibility determining and framework regions (CDR and FR), which are essential for recognizing the antigen. Alterations in these structures may affect the antibody’s ability to recognize its antigen and reduce its efficacy.
Currently, methods like UV and fluorescent spectroscopy are used for tertiary structure analysis, while Fourier Transform Infrared (FTIR) and CD spectroscopy are used for secondary structure analysis. However, X-ray crystallography and NMR spectroscopy yield much more accurate and detailed structural information. These are discussed in detail in our next post. We will also discuss the experimental approach used by the SARomics team, which, with high cost-efficiency, combines X-ray crystallography and NMR spectroscopy for high-precision comparability analysis of monoclonal antibody HOS.
Follow us on LinkedIn to get updates on our future articles. For project discussions, please see our antibody structure services page. We invite you to visit our publications page for examples of projects we have worked on.