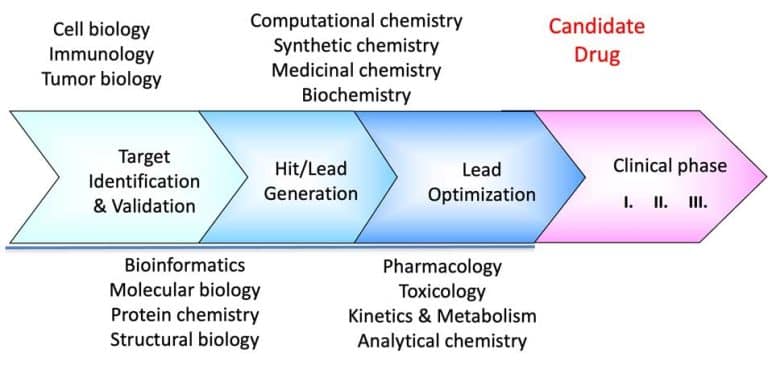
Room-Temperature Crystallography In Drug Design
In this post we want to bring to the attention of our readers the revival of room-temperature (RT) protein crystallography using synchrotron radiation and its potential applications in structure-based drug design to study of functionally-important conformations.
After more than two decades of cryo-crystallography, finding someone with skills in mounting protein crystals for room-temperature X-ray crystallography won’t be easy. It would be safe to say that most young crystallographers today haven’t even seen an X-ray capillary of the type used in RT crystallography. Robert Steiner has put together a virtual issue of Acta Cryst. D, Structural Biology, with articles on room-temperature crystallography.
Why room-temperature X-ray crystallography?
An obvious advantage of doing room-temperature crystallography is, as stated at the site of MiTeGen. This company offers products and tools for X-ray crystallography:” Roughly 98% of low-temperature data sets collected on structural biology beamlines do not yield diffraction data of sufficient quality to determine a structure. Room temperature measurements are essential to determine whether the cause of poor diffraction is poor as-grown crystal quality, sample damage caused by ligand and/or cryoprotectant soaks, and/or sample damage caused by the flash cooling process. Far too much effort is wasted searching for cryoprotection conditions for crystals that are poorly ordered to start with.”
Other shortcomings of cryo-crystallography are listed in a review article by Robert E. Thorn in the issue mentioned above:
- Regions of ordered density in a cryo-structure may be disordered at room temperature (and vice versa), and disorder at room temperature may be of functional relevance. However, this is missed in cryo-crystallography since most structures are refined to a single conformation. Room-temperature data collection can facilitate the identification of functionally important alternative conformations at active sites and elsewhere.
- The cryoprotectant used in cryo-crystallography may perturb side chain conformations and contribute additional electron density, including within the active site region.
- Cryo-cooling may degrade crystal lattice order, even if the thermal disorder is reduced. It may also introduce crystal non-isomorphism and high mosaicity. Variability in cryoprotectant soaks and cooling may also result in non-isomorphism.
These realizations and the recent technical developments discussed in Robert E. Thorn’s review together triggered the revival of room-temperature X-ray crystallography.
- The development of X-ray-free electron laser (XFEL) sources and serial crystallography at synchrotrons
- , as well as developing serial-sample delivery and software for processing and modeling data collected from many tiny crystals.
- Improved detector speed and sensitivity; new techniques for time-resolved study of reactions and conformational dynamics within crystals.
Conformational diversity and ligand binding
The new findings show that room-temperature and multi-temperature crystallography can be used to study functionally important conformations and identify temperature-dependent differences in protein-ligand interactions, such as new binding sites and unique binding poses. Such studies may provide a more complete understanding of protein-ligand interactions, making room-temperature crystal structures a better representation of the functionally relevant conformations of proteins and, thus, more efficient for drug discovery.
An example of a study of ligand-binding discrepancies between cryo-cooled and RT-temperature structures is in the work by Huang and co-workers, who used a temperature-variation approach to examine the binding of TL00150, a 175.15 Da endothiapepsin inhibitor. The authors report the details of the experimental setup for the temperature-dependent measurements and show that at 298 K, depending on DMSO concentration, the fragment TL00150 had two binding positions. In contrast, at cryo-temperature (100 K), it was only observed in a single position at the S1 part of the active site pocket. Subsequently, data for a series of structures with bound ligands were collected to assess ligand occupancy at different temperatures. The data showed different ligand binding states with different occupancies at the S1 and S1′ sites. This was explained by a flexible loop close to the protein’s active site.
The new technical developments, by making room-temperature X-ray crystallography and multi-temperature measurements relatively easily accessible at synchrotrons, provide new opportunities for studying functionally important conformations that may help the drug design process. Last but not least, we could start testing crystals directly at synchrotrons at room temperature without all the hassle of finding a suitable cryoprotectant and freezing!
Follow our publications on LinkedIn. For project discussion, please visit our X-ray crystallography services page.